



文献解读 | 一种基于CNV-seq和STR-seq结合的检测引起自然流产的非整倍体及染色体异常来源
发布日期:2023-03-22

近日,复旦大学附属妇产科医院上海集爱遗传与不育诊疗中心雷彩霞医师和复旦大学生命科学学院卢大儒教授实验室的廖凯在临床医学杂志 Journal of Clinical Medicine(IF:4.964)上共同发表了一篇题为《A Novel System for the Detection of Spontaneous Abortion-Causing Aneuploidy and Its Erroneous Chromosome Origins through the Combination of Low-Pass Copy Number Variation Sequencing and NGS-Based STR Tests》的文章。复旦大学附属妇产科医院上海集爱遗传与不育诊疗中心副所长孙晓溪和韦翰斯生物的CTO杨敬敏博士为文章的共同通讯作者。研究提出了一种基于CNV-seq和STR-seq结合检测引起自然流产的非整倍体及染色体异常来源方的新方法,对未来的流产物检测及染色体异常来源判断具有深远的意义。
背景介绍
早期流产是妊娠失败的主要原因之一,除了内分泌和解剖学异常、获得性血栓形成倾向或诱发自然流产的环境因素外,染色体是决定胚胎命运的重要因素。胚胎染色体异常导致超过50%的早孕期流产,其中染色体三体、单体和三倍体是流产样本中最常见的染色体异常类型明确导致流产的原因,可以指导流产夫妇再次怀孕或进行医疗干预。
在过去的几十年里,从最经典的细胞遗传核型分析,荧光原位杂交(FISH),到比较基因组杂交芯片(aCGH)和单核苷酸多态性微阵列芯片(SNParray),定量荧光聚合酶链反应(Q-PCR)和商用多重引物连接扩增(MLPA)试剂盒,再到基于低分辨全基因组测序的拷贝数变异测序(CNV-seq),流产物分析技术的分辨率越来越高,操作上更简单便捷,结果也变得越来越准确。然而,上述方法均不能鉴别异常染色体的来源,例如单体、三体及三倍体来源。例如“69,XXX”和“69,XXY”等。
研究概述
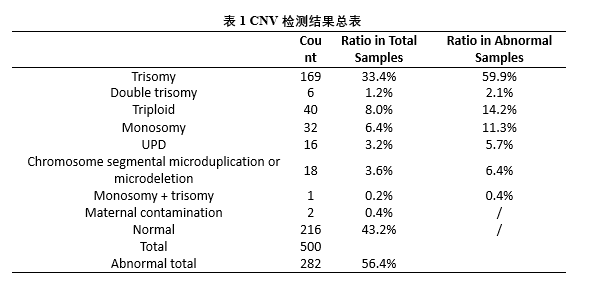
为了解决上述问题,本研究在22条常染色体和两条性染色体(X和Y染色体)上共开发了386个STR位点,将基于NGS的STR检测与CNV-seq结合用于流产样本的分析。与G显带核型分析相比,该系统在500例不明原因的复发性自然流产中,将流产样本中的染色体异常检出率提高到56.4%。在检测到的非整倍体异常中,最常检测到的是三体异常(33.4%,染色体异常总共组为59.9%)。在三体样本中,94.7%的额外染色体来自母系,5.3%来自父系。此外,本研究中还发现了9个较罕见的全基因组UPD样本和6个双三体样本,STR结果表明全基因组UPD样本中8个为父系全基因组UPD,双三体样本有2个样本的两条额外染色体分别来自母系和父系。
图1 各染色体STR 基因座的分布图
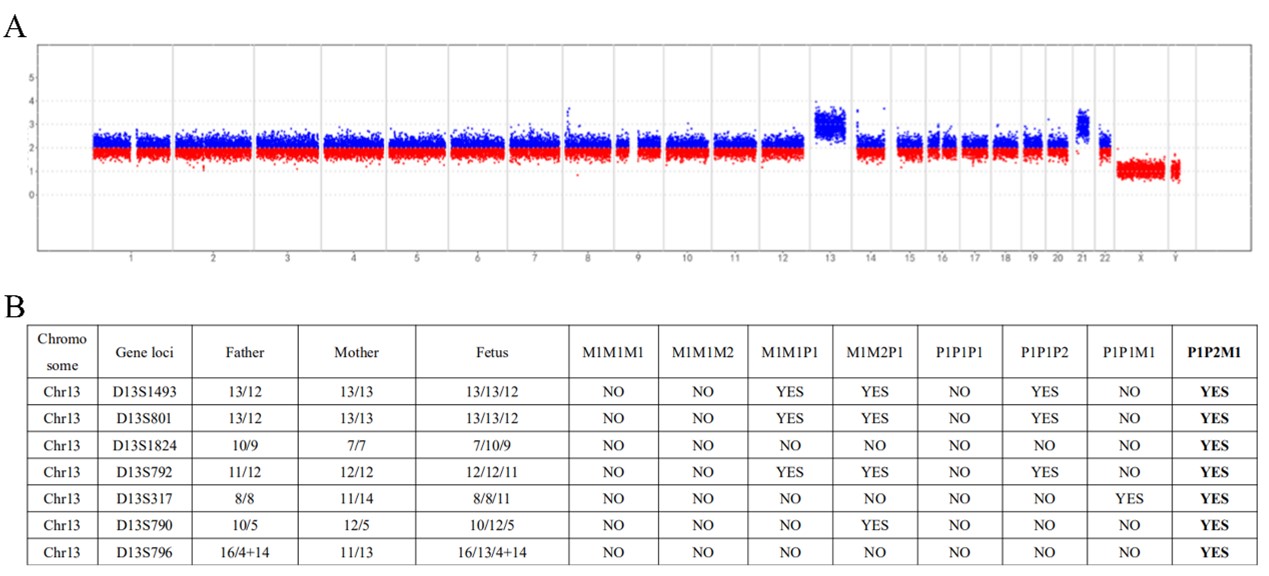
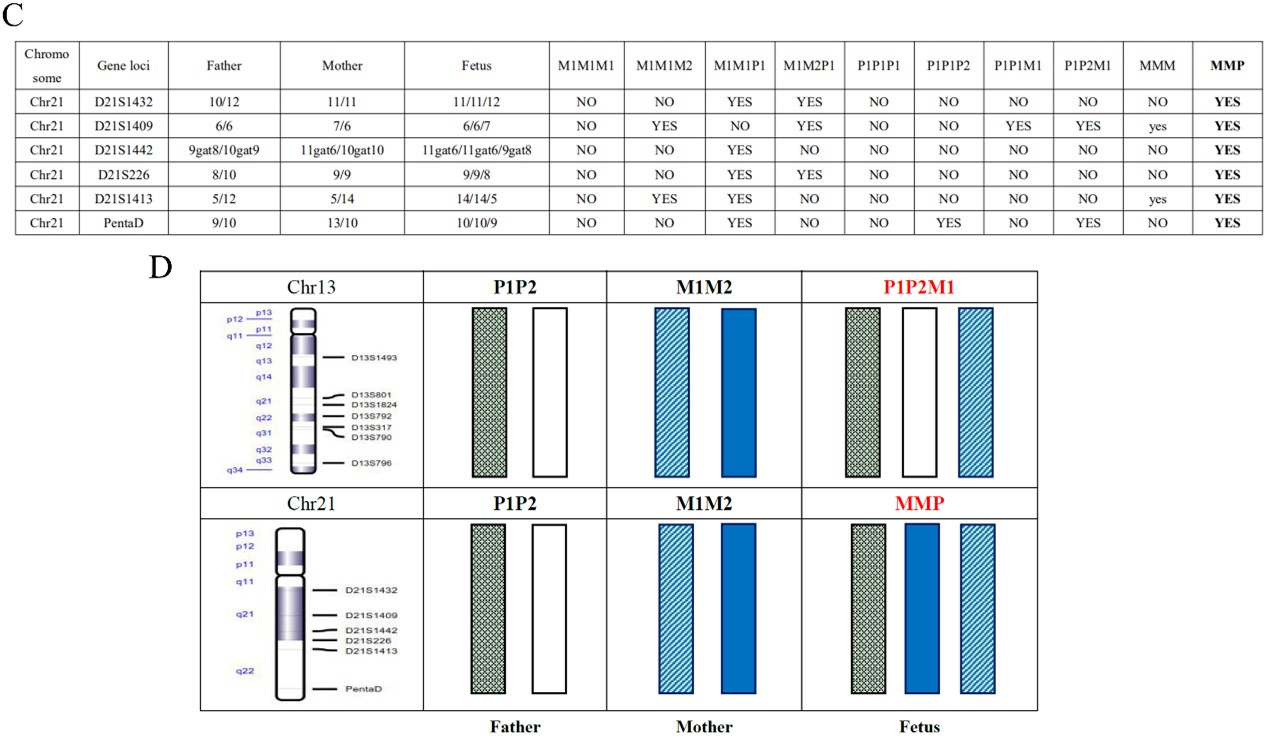
图2 双三体样本结果示例
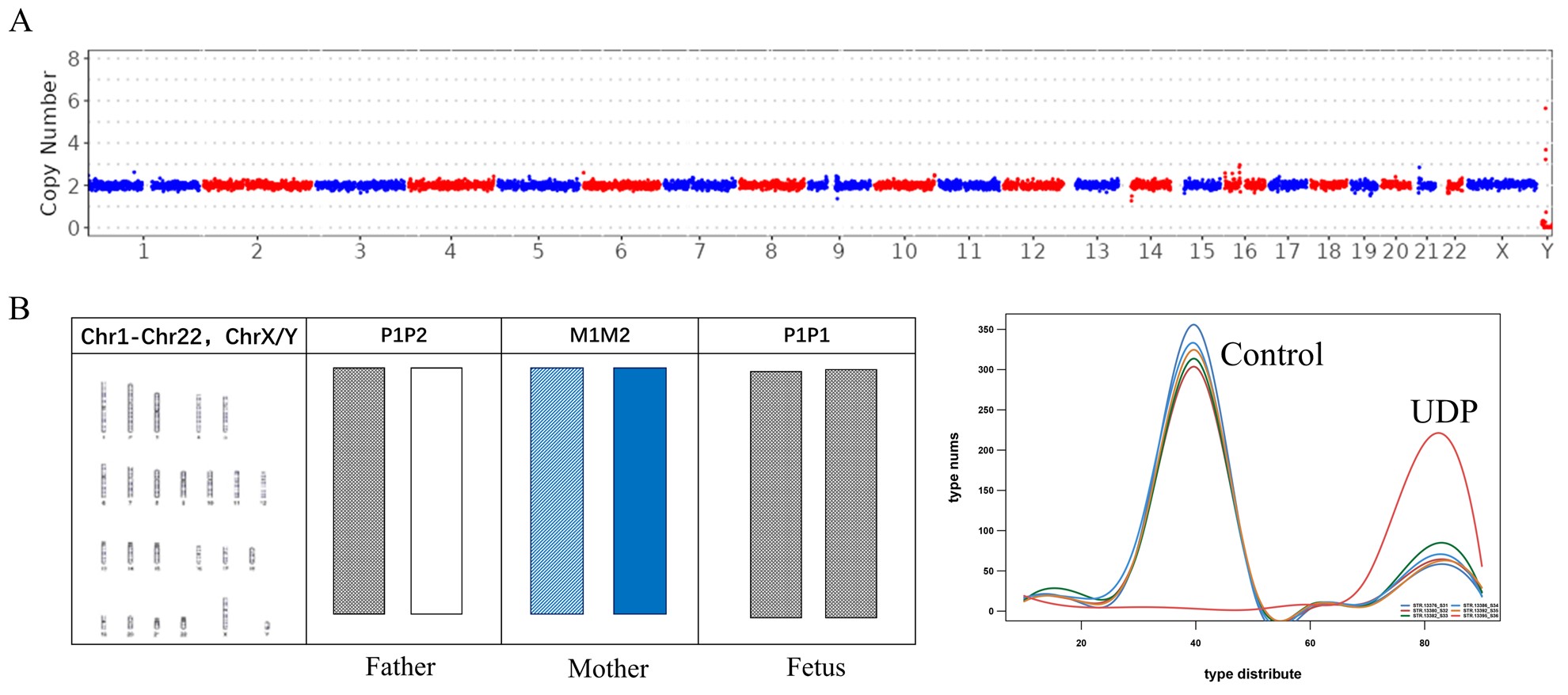
图3 单亲二倍体样本结果示例
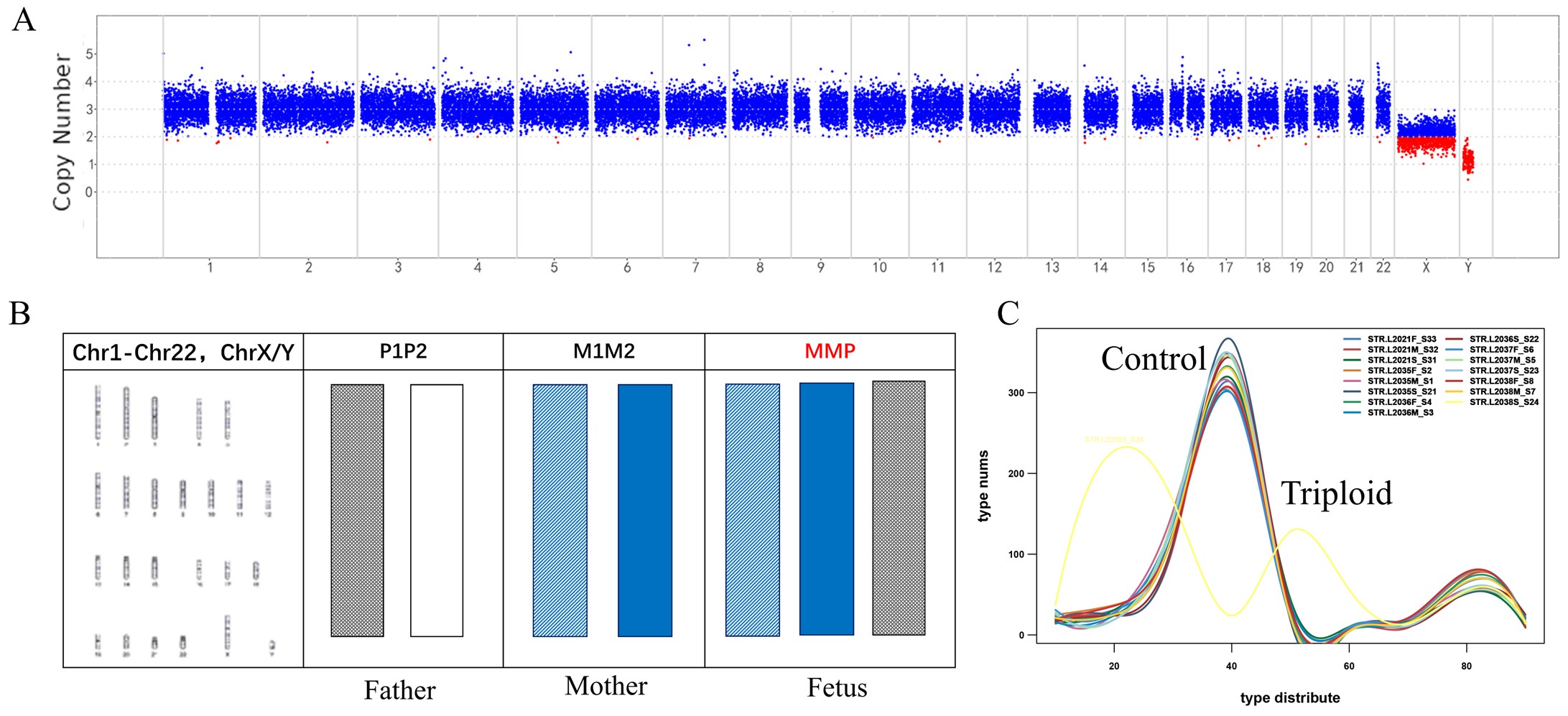
图4 三倍体样本结果示例
研究小结
与传统核型分析相比,该方法不仅提高了复发性流产夫妇流产样本中染色体异常的检出率,而且能够追溯异常染色体的亲本来源,同时能够有效检测三倍体和UPD,并识别流产样本中的母源细胞污染。此外,因同源重组决定了配子细胞中的等位基因与其亲本的等位基因并不完全相同,在三体组中,STR检测到的染色体的单倍型与结果中其他亲本染色体的单倍型可推测染色体异常发生的细胞周期,但是因为着丝粒周围区域的STR不包括在该panel中,暂时无法推测错误发生在减数分裂的具体阶段。该系统适用于范围广泛的样品,包括血液、组织、唾液、细胞和石蜡,此外它还具有成本更低、检测周期更短的优势。未来,该方法将继续改进,尝试将其应用到辅助生殖技术中,帮助医生快速鉴定自然流产胚胎的核型,决定是否进行PGT-A检测,为临床妊娠指导提供了更多的参考信息。
微信扫一扫
关注该公众号